1.2 Genome sequencing
1.2.1 DNA sequencing
Several methods for DNA sequencing exist, among them the chain termination method first developed by (Sanger, Nicklen, and Coulson 1977) is the most popular but alternative techniques such as chemical degradation sequencing (Maxam and Gilbert 1977) and pyrosequencing (Nyrén, Pettersson, and Uhlén 1993) are also used.
Chain termination method is based on the principle that single-stranded DNA molecules that differ in length by just a single nucleotide can be separated by polyacrylamide gel electrophoresis1. This procedure is illustrated and explained in Figure 1.2.
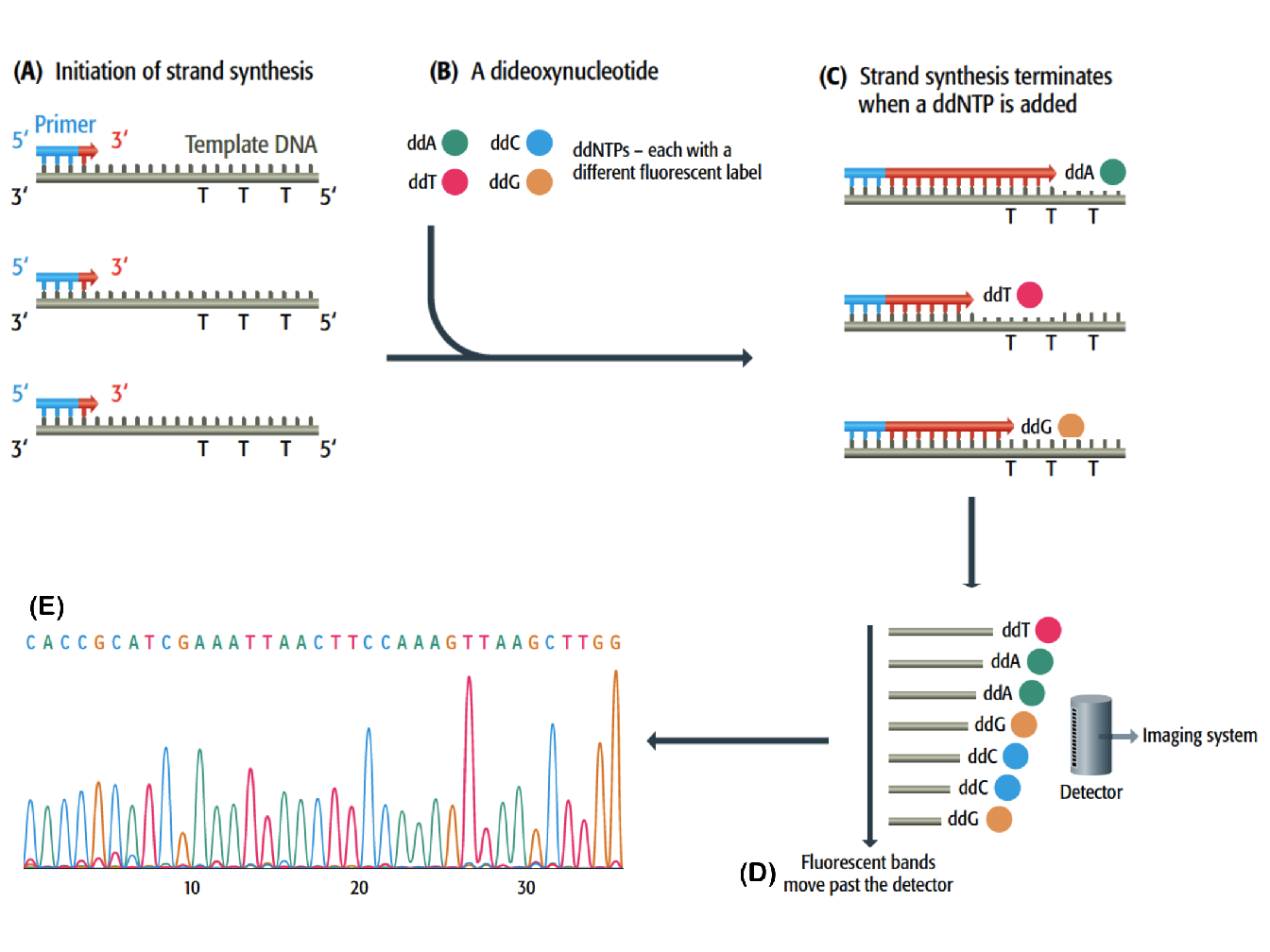
Figure 1.2: Chain termination DNA sequencing (Brown et al. 2007). (A) Use of universal primers for the synthesis of DNA complementary of a single-stranded template. (B) Incorporation of small amount of fluorescent dideoxynucleotides (ddATP, ddTTp, ddCTP and ddGTP), each with a different fluorescent label. (C) The ddNTP block the synthesis of DNA because they have a hydrogen atom rather than a hydroxyl group attached to the 3’ carbon. (D) Each labelled DNA strand passes through a polyacrylamide gel electrophoresis, migrating more or less according to their length, and after separation a fluorescent detector is capable of discriminating the labels attached to the ddNTP. (E) The information is passed to the imaging system and a sequence of DNA is printed out. The sequence is represented by a series of peaks, one for each nucleotide position.
Pyrosequencing is a method generally used for the rapid determination of very short sequence of DNA and does not required electrophoresis or any fragment separation procedure as with chemical degradation sequencing. Since it can only generate a few tens of base pairs per experiment, it is used when many short sequences must be generated as fast as possible, for instance in single-nucleotide polymorphism typing. With this technique, the template is copied in a straightforward manner without added ddNTP and, as the new strand is being made, the order in which the deoxynucleotide are incorporated can be followed (see Figure 1.3 for more details).
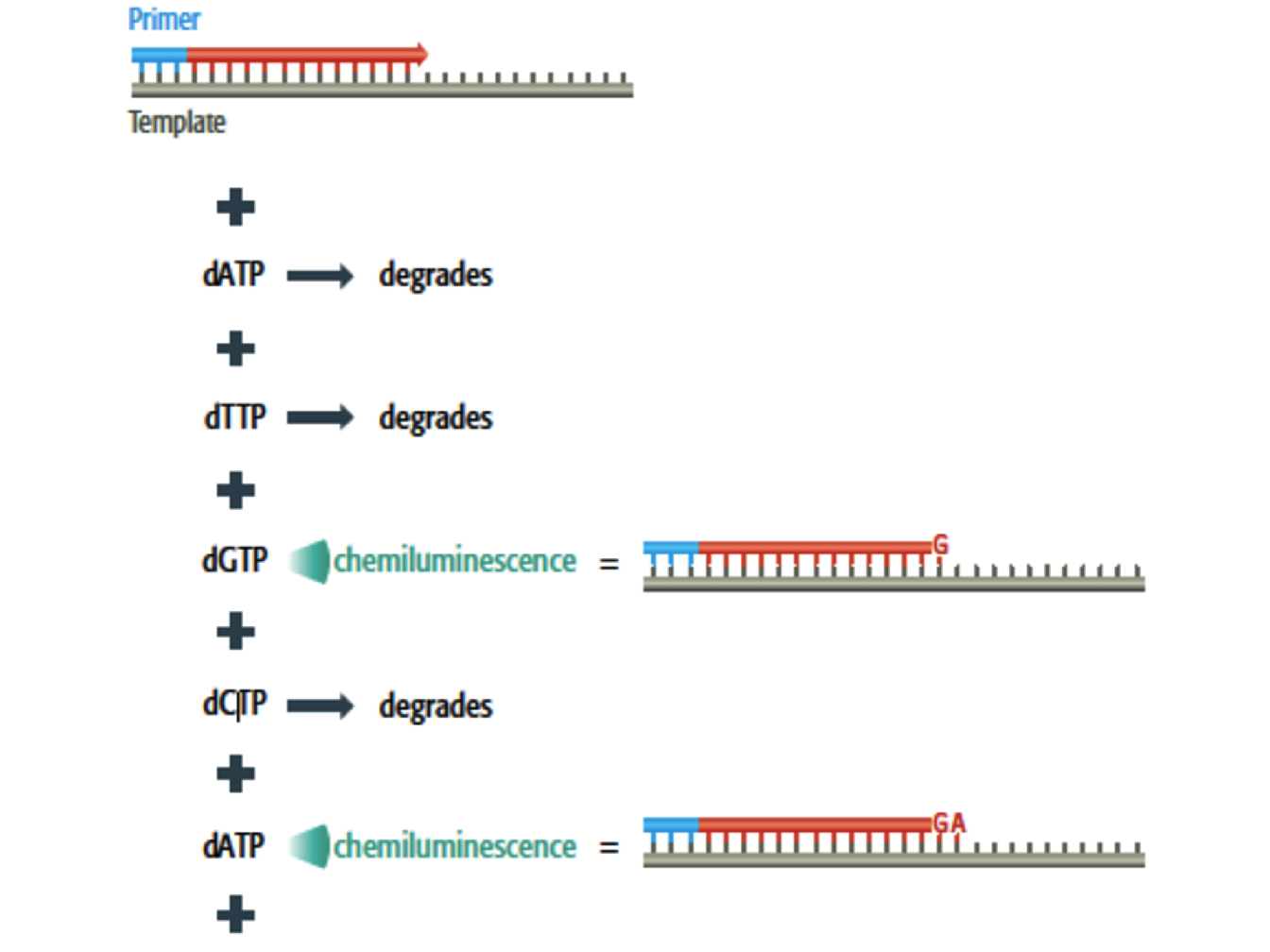
Figure 1.3: Pyrosequencing (Brown et al. 2007). Each deoxynucleotide is added individually, along with a nucleotidase enzyme that degrades the deoxynucleotide if it is not synthesized. The incorporation is detected by a flash of chemiluminescence induced by the pyrophosphate released from the deoxynucleotide. The order in which the deoxynucleotide are added to the growing strand can therefore be followed.
1.2.2 Sequence assembly
One of the main challenges in genome sequencing is to master the assembly of the multitude of short sequences generated by DNA sequencing techniques in order to reconstruct the complete continuous sequence of chromosome that can reach a length of several tens of megabases. The most straightforward method to sequence assembly is to build up the master sequence by directly searching for overlaps between all the short sequences. This method is known as the shotgun method (Anderson 1981). The shotgun method is the standard approach for sequencing small prokaryotic2 genome but it is not suited to the analysis of larger genome because the required data analysis becomes too complex as the number of fragment increases (for \(n\) fragments, the number of possible overlaps is \(2n^2 - 2n\)). Moreover it can lead to errors when repetitive regions of a genome are analysed because when a repetitive sequence is broken into fragments, many of the resulting pieces contain the same sequence motifs.
To overcome these issues, techniques that make use of a genome map to guide the assembly are used, namely the whole-shotgun method and clone contig method (Figure 1.4):
Whole-genome shotgun method. This method takes the same approach as the standard shotgun procedure but uses the distinctive features on the genome map as landmark to assemble the whole sequence. Reference to the map ensures that regions containing repetitive DNA are assembled correctly.
Clone contig method. In this method the genome is broken into manageable segments which are short enough to be assembled accurately by the shotgun method. Once the sequence of a segment has been completed, it is positioned at its correct location on the map
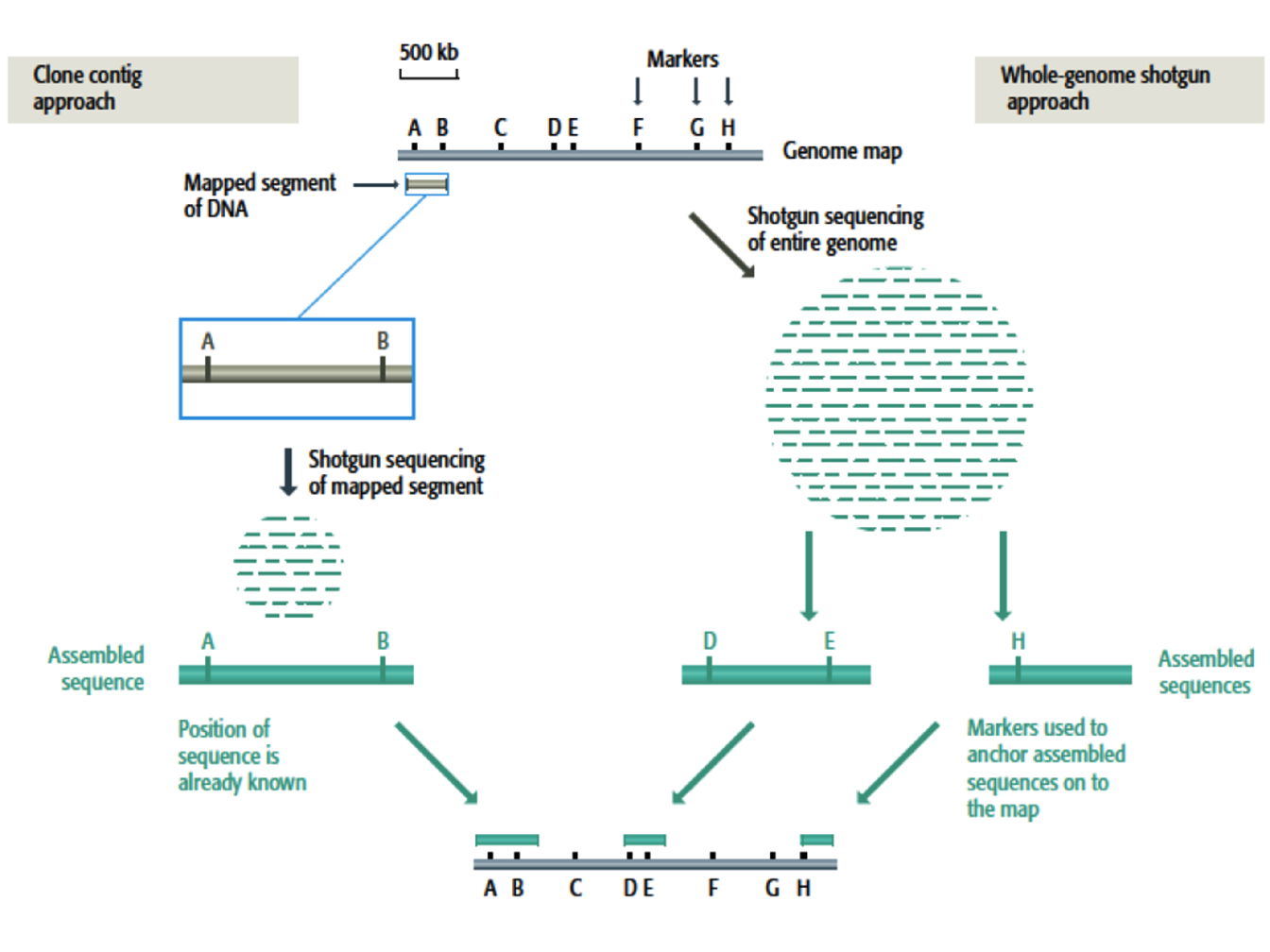
Figure 1.4: Clone contig and whole-genome shotgun for sequence assembly (Brown et al. 2007). To illustrate both techniques, a genome map of linear DNA molecule of 2.5 Mb has been represented together with the location of 8 known markers(A-H). On the left, the clone contig approach starts with a segment of DNA whose position on the genome is known since it contains the markers A and B. The segment is sequenced by the shotgun method and the master sequence placed at its known position on the map. On the right, the whole-genome shotgun method involves random sequence of the entire genome resulting in pieces of contiguous sequence. If a contiguous sequence contains a marker then it can be positioned on the map.
References
Anderson, Stephen. 1981. “Shotgun Dna Sequencing Using Cloned Dnase I-Generated Fragments.” Nucleic Acids Research 9 (13): 3015–27.
Brown, T.A., D.B.S.T. Brown, T.A. Brown, and L.B.T. Brown. 2007. Genomes 3. Taylor & Francis Group, an Informa Business. Garland Science Pub. https://books.google.fr/books?id=Cjl98tqp6rsC.
Maxam, Allan M, and Walter Gilbert. 1977. “A New Method for Sequencing Dna.” Proceedings of the National Academy of Sciences 74 (2): 560–64.
Nyrén, Pettersson, Bertil Pettersson, and Mathias Uhlén. 1993. “Solid Phase Dna Minisequencing by an Enzymatic Luminometric Inorganic Pyrophosphate Detection Assay.” Analytical Biochemistry 208 (1): 171–75.
Sanger, Frederick, Steven Nicklen, and Alan R Coulson. 1977. “DNA Sequencing with Chain-Terminating Inhibitors.” Proceedings of the National Academy of Sciences 74 (12): 5463–7.
Polyacrylamide gel electrophoresis (PAGE) is a technique widely used in genetics to separate biological macromolecules, sush as nucleic acids, according to their electrophoretic mobility.↩
A prokaryote is a unicellular organism that lacks a membrane-bound nucleus, mitochondria, or any other membrane-bound organelle.↩